Molecular’s ETB Platform Technology
Although chemotherapy remains the cornerstone of treatment for most cancers, the advent of new and targeted classes of therapies has dramatically changed outcomes in the treatment of disease. The advent of monoclonal antibodies, signal transduction inhibitors and, most recently, immuno-oncologics have provided substantial clinical benefit in both the relapsed and refractory setting and, when used in combinations, in earlier lines of therapy. Molecular believes that ETBs represent a new class of targeted agents with differentiated biology that are well-positioned to improve outcomes in cancer patients.
ETBs appear to induce the internalization of non- or poorly-internalizing targets, have a differentiated mechanism of action (enzymatic and irreversible ribosome inactivation), have relatively predictable PK profiles and can be readily manufactured to cGMP standards. From a library of antibody-like targeting domains, Molecular’s research and design platform allows for the comprehensive in vitro selection of a lead ETB to a given target based on affinity and specificity, potency and expression. Lead selection is confirmed through the use of animal models to verify PK, absorption, distribution, metabolism and excretion, and potency. ETBs possess potent direct cell killing effects via a differentiated mechanism of action, can force receptor internalization, and can be used to deliver payloads such as foreign class I antigen to the cytosol.
In all clinical-stage ETBs, Molecular utilizes a highly potent and proprietarily de-immunized SLTA scaffold that elicits significantly reduced innate and adaptive immunogenic responses as demonstrated in preclinical and animal studies. For indications where tumors have been demonstrated to be sensitive to T-cell engagement, Molecular has developed ETBs that deliver foreign class I viral antigens for presentation on the surface of the tumor: Molecular’s Antigen Seeding Technology, a differentiated approach to immuno-oncology. Molecular has integrated its Antigen Seeding Technology into the PD-L1 targeting ETB, MT-6402, and continues to build out animal models to further validate and screen additional ETB candidates to support this approach.
Molecular believes that its proprietary ETB technology platform represents a differentiated approach in oncology. ETBs possess the targeting specificity of antibody-based therapeutic approaches but deliver highly potent payloads that disrupt protein synthesis, a fundamental function of a cancer cell, in a manner not subject to traditional chemotherapy resistance mechanisms or target internalization limitations, as with ADCs. Molecular is also seeking to expand the universe of potential targets subject to pharmaceutical treatments by exploiting the ETB’s ability to force internalization against receptors that do not normally internalize.
Novel mechanisms of action are needed in oncology treatment, and Molecular believes that its ETB platform technology’s differentiated mechanisms of action may offer unique benefits over existing treatment modalities.
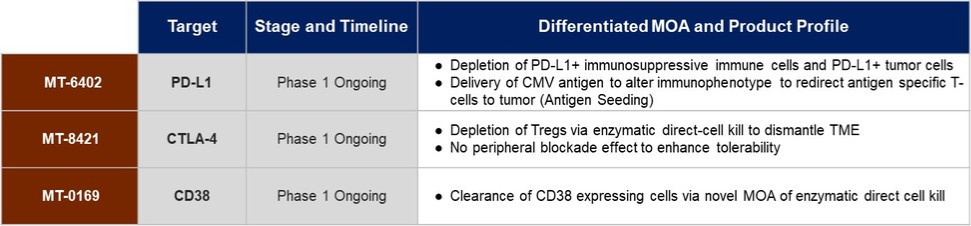
ETB Product Pipeline
Molecular is developing a pipeline of ETBs that Molecular believes will have the ability to provide a meaningful and long-lasting benefit to cancer patients. Molecular plans to develop each of these as single agents and/or in combination with other therapies, as applicable. The following table depicts Molecular’s current pipeline:
9